Calculating Acceleration: Instant Center of Rotation in Plane Motion
Not what you're looking for?
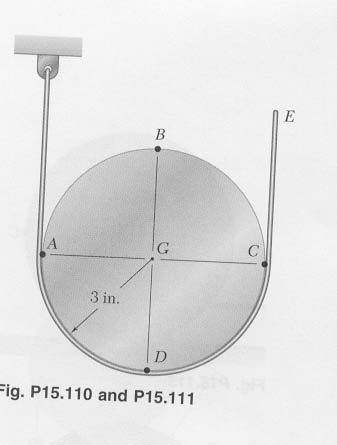
Question: The motion of the 3-in.-radius cylinder is controlled by the cord shown (please refer to attachment for diagram). Knowing that end E of the cord has a velocity of 12 in./s and an acceleration of 19.2 in./s^2, both directed upward, determine the acceleration (a) of point A, (b) of point B.
{kind=link}
Purchase this Solution
Solution Summary
This solution is comprised of a step-by-step guide on how to approach and work through the formulas required to calculate acceleration as it pertains to this question. A jpeg attachment is also provided to illustrate a schematic diagram for this solution, which may help to provide further clarification.
Solution Preview
a.) Because, point E is moving upward with a speed of 12 in/s,
The point G will move upward with a speed = 12/2 = 6 in/s
While the cylinder's point A will have a speed 0 in/s downward, therefore, it will have a speed of 6 ...
Education
- BEng, Allahabad University, India
- MSc , Pune University, India
- PhD (IP), Pune University, India
Recent Feedback
- " In question 2, you incorrectly add in the $3.00 dividend that was just paid to determine the value of the stock price using the dividend discount model. In question 4 response, it should have also been recognized that dividend discount models are not useful if any of the parameters used in the model are inaccurate. "
- "feedback: fail to recognize the operating cash flow will not begin until the end of year 3."
- "Answer was correct"
- "Great thanks"
- "Perfect solution..thank you"
Purchase this Solution
Free BrainMass Quizzes
Basic Physics
This quiz will test your knowledge about basic Physics.
Introduction to Nanotechnology/Nanomaterials
This quiz is for any area of science. Test yourself to see what knowledge of nanotechnology you have. This content will also make you familiar with basic concepts of nanotechnology.
Intro to the Physics Waves
Some short-answer questions involving the basic vocabulary of string, sound, and water waves.
Classical Mechanics
This quiz is designed to test and improve your knowledge on Classical Mechanics.
The Moon
Test your knowledge of moon phases and movement.