Intersect of Surface and Line in 3D Space
Not what you're looking for?
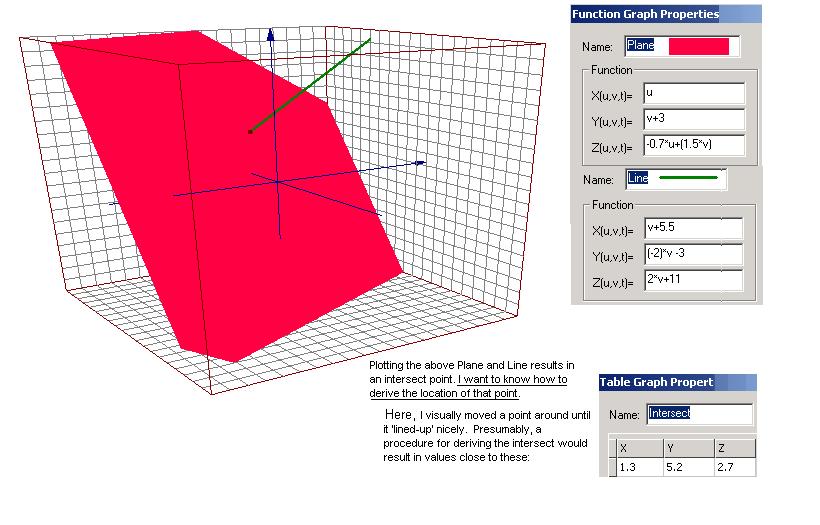
Attached, find a graphic of a Plane and a Line.
I'm hoping the solution can be conveyed without Matrix math, and am thinking it ought to be solvable using 'conventional' simutaneous equations (which I almost 'get').
Too, I'm hoping the solution will be fairly 'general', in the sense that, after understanding this first effort, that I will be able to apply it to a more complex surface.
THANKS!
{kind=link}
Purchase this Solution
Solution Summary
A solution to the intersect of a line and a surface is found using a substitution method.
Solution Preview
Hello,
I appreciate your work in visualizing the plane and line in space and obtaining the approximate point of intersection.
In the attached file, I have explained the complete procedure (substitution method, not matrix method)of obtaining the required point of intersection. I hope you can understand this.
Well. The solutions can be obtained using a simple substitution method.
I appreciate your work in getting ...
Purchase this Solution
Free BrainMass Quizzes
Know Your Linear Equations
Each question is a choice-summary multiple choice question that will present you with a linear equation and then make 4 statements about that equation. You must determine which of the 4 statements are true (if any) in regards to the equation.
Solving quadratic inequalities
This quiz test you on how well you are familiar with solving quadratic inequalities.
Probability Quiz
Some questions on probability
Graphs and Functions
This quiz helps you easily identify a function and test your understanding of ranges, domains , function inverses and transformations.
Exponential Expressions
In this quiz, you will have a chance to practice basic terminology of exponential expressions and how to evaluate them.